
LIVMARLI 9,5 mg-mL, solution buvable, boîte de 1 flacon ( 3 seringues pour administration orale) de 30 ml
Dernière révision : 28/06/2024
Taux de TVA : 2.1%
Prix de vente : 24 456,38 €
Taux remboursement SS : 65%
Base remboursement SS : 24 456,38 €
Laboratoire exploitant : MIRUM PHARMA FRANCE
Source :

Livmarli est indiqué pour le traitement :
- du prurit cholestatique chez les patients atteints du syndrome d'Alagille à partir de l'âge de 2 mois,
- de la cholestase intrahépatique progressive familiale (PFIC) chez les patients âgés de 3 mois et plus.
Hypersensibilité à la substance active ou à l'un des
excipients mentionnés à la rubrique Liste des excipients.
Insuffisance hépatique et/ou rénale sévère chez les patients atteints de PFIC, en raison du risque potentiel de toxicité liée au propylène glycol utilisé comme excipient (voir rubrique Mises en garde spéciales et précautions d'emploi).
Le maralixibat agit en inhibant le transporteur iléal des acides biliaires (IBAT, ileal bile acid transporter) et en interférant avec le cycle entéro-hépatique des acides biliaires. Par conséquent, les affections, les médicaments ou les interventions chirurgicales altérant la motilité gastro-intestinale ou le cycle entéro-hépatique des acides biliaires, y compris le transport des sels biliaires vers les canalicules biliaires, sont susceptibles de réduire l'efficacité du maralixibat.
De ce fait, les patients atteints de PFIC2 chez lesquels la protéine BSEP (pompe d'export des sels biliaires) est totalement absente ou dysfonctionnelle (c.-à-d. les patients atteints du sous-type BSEP3 de la PFIC2) ne devraient pas répondre au maralixibat.
La diarrhée a été décrite comme un effet indésirable très fréquent lors de la prise de maralixibat (voir rubrique Effets indésirables). La diarrhée peut conduire à une déshydratation. Les patients doivent faire l'objet d'une surveillance régulière au cours des épisodes de diarrhée afin d'assurer une hydratation suffisante.
Les patients présentant une diarrhée chronique nécessitant une réhydratation intraveineuse ou une intervention nutritionnelle n'ont pas été étudiés dans le cadre des essais cliniques.
Une augmentation des ALAT et ASAT a été observée chez certains patients traités par le maralixibat (rubrique Effets indésirables). Le bilan hépatique des patients devra être surveillé avant et pendant le traitement par le maralixibat.
Il est recommandé de mesurer les taux de vitamines liposolubles (VLS) (vitamines A, D, E) et le rapport normalisé international (INR) chez tous les patients, avant d'instaurer le traitement par Livmarli, et de les surveiller conformément aux pratiques cliniques usuelles. Si un déficit en VLS est diagnostiqué, un traitement de complémentation devra être prescrit.
Chez les patients atteints de PFIC dont la capacité à métaboliser et/ou éliminer le propylène glycol est altérée (par exemple, ceux atteints d'insuffisance hépatique et/ou rénale et les patients âgés de moins de 5 ans), le risque de toxicité du propylène glycol est accru lorsque des doses élevées de Livmarli sont administrées. Une réduction de la dose de Livmarli est recommandée chez ces patients (voir rubrique Posologie et mode d'administration et rubrique Mises en garde spéciales et précautions d'emploi sous « Propylène glycol et risque potentiel de toxicité ») ; les patients atteints de PFIC et présentant une insuffisance hépatique et/ou rénale sévère ne doivent pas être traités par Livmarli (voir rubrique Contre-indications).
Excipients à effet notoire :
Propylène glycol et risque potentiel de toxicité
Ce médicament contient 364,5 mg de propylène glycol (E 1520) par mL de solution buvable.
Syndrome d'Alagille : en cas d'administration de Livmarli à la dose de 380 microgrammes/kg une fois par jour, l'exposition au propylène glycol pourra atteindre jusqu'à 17 mg/kg/jour.
PFIC : en cas d'administration de Livmarli à la dose de 285 microgrammes/kg deux fois par jour, l'exposition au propylène glycol pourra atteindre jusqu'à 26 mg/kg/jour et à la dose de 570 microgrammes/kg de Livmarli deux fois par jour, l'exposition au propylène glycol pourra atteindre jusqu'à 50 mg/kg/jour.
La quantité totale de propylène glycol contenue dans tous les médicaments et les compléments alimentaires, y compris dans la solution buvable de Livmarli, doit être prise en compte lors de l'évaluation du risque potentiel de toxicité liée au propylène glycol, en particulier chez les patients ayant une capacité limitée à métaboliser ou excréter le propylène glycol (par exemple, les patients âgés de moins de 5 ans ou ceux dont la fonction rénale ou hépatique est altérée) (voir rubriques Posologie et mode d'administration et Contre-indications). L'administration concomitante avec n'importe quel substrat pour l'alcool déshydrogénase comme l'éthanol peut augmenter le risque de toxicité liée au propylène glycol.
Les effets indésirables liés à la toxicité potentielle du propylène glycol incluent par exemple : hyperosmolalité (avec ou sans acidose lactique), dysfonction rénale (nécrose tubulaire aiguë), insuffisance rénale aiguë, cardiotoxicité (arythmie, hypotension) ; troubles du système nerveux central (dépression, coma, convulsions), dépression respiratoire, dyspnée ; dysfonction hépatique, réaction hémolytique (hémolyse intravasculaire) et hémoglobinurie ; ou dysfonctionnement organique multisystémique. Les signes et symptômes d'une possible toxicité du propylène glycol devront être surveillés chez les patients.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.‑à‑d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Plus de 280 patients atteints de maladies hépatiques cholestatiques et âgés de 1 mois à 24 ans ont été traités par le maralixibat au cours d'études cliniques en aveugle et en ouvert, dont 94 patients atteints du syndrome d'Alagille traités sur une période allant jusqu'à 5 ans et 134 patients atteints de PFIC traités sur une période allant jusqu'à 7 ans.
Le profil de sécurité du maralixibat est cohérent dans les différentes indications et les différents groupes d'âge. L'effet indésirable rapporté le plus fréquemment chez les patients atteints du syndrome d'Alagille et âgés de plus de 12 mois est la diarrhée (36,0 %), suivie par les douleurs abdominales (29,1 %). Chez les patients atteints de PFIC et âgés de plus de 12 mois, les effets indésirables les plus fréquents ont été également la diarrhée (27,7 %) et les douleurs abdominales (6,4 %). L'effet indésirable rapporté le plus fréquemment chez les patients atteints du syndrome d'Alagille et âgés de moins de 12 mois est la diarrhée (20,0 %). La diarrhée (23,5 %) était également l'effet indésirable rapporté le plus fréquemment chez les patients atteints de PFIC et âgés de moins de 12 mois.
Tableau récapitulatif des effets indésirables
Pour le syndrome d'Alagille, le profil de sécurité du maralixibat a été établi sur la base de l'analyse groupée des données issues de 5 études cliniques menées chez des patients (n = 86) âgés de 1 à 17 ans (âge médian : 5 ans). La durée d'exposition médiane était de 2,5 ans (intervalle : 1 jour à 5,5 ans). Pour la PFIC, le profil de sécurité est principalement basé sur l'analyse des données comparées au placebo, en double aveugle, issues de l'étude pivot sur la PFIC et de l'étude d'extension en ouvert (n = 93, dont 88 patients traités par le maralixibat à la dose recommandée). Les patients traités par le maralixibat étaient âgés de 1 à 17 ans (âge médian : 4 ans). La durée d'exposition médiane était de 83,5 semaines (intervalle : 1,7 à 177,1 semaines). Des données supplémentaires concernant la sécurité à long terme à une plus faible dose de maralixibat (≥ 266 microgrammes/kg/jour) ont été collectées au cours d'une étude clinique de phase 2 (LUM001‑501) et d'une étude de suivi à long terme en ouvert (MRX‑800 ; durée d'exposition totale allant jusqu'à 7 ans).
Dans le groupe d'âge des patients âgés de moins de 1 an, 17 patients atteints du syndrome d'Alagille et 10 patients atteints de PFIC ont été traités par le maralixibat aux doses recommandées (voir rubrique Propriétés pharmacodynamiques).
Le tableau 3 présente les effets indésirables rapportés d'après ces analyses.
Les effets indésirables survenus chez les patients traités par le maralixibat sont présentés ci-dessous par classe de système d'organe MedDRA et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 3 : Effets indésirables rapportés chez les patients atteints du syndrome d'Alagille et de PFIC
|
Classe de système d'organe |
Fréquence |
Effets indésirables |
|
Affections gastro-intestinales |
Très fréquent |
Diarrhée |
|
Douleurs abdominales |
||
|
Affections hépatobiliaires |
Fréquent |
ALAT et ASAT augmentées |
Description de certains effets indésirables
Tous les cas de diarrhée signalés ont été de sévérité légère à modérée ; un épisode sévère de douleurs abdominales a été rapporté chez 1 patient atteint du syndrome d'Alagille. Aucun des épisodes de diarrhée ou de douleurs abdominales n'a été grave. Dans la majorité des cas, la diarrhée et les douleurs abdominales sont apparues au cours du premier mois de traitement. Pour le syndrome d'Alagille comme pour la PFIC, la durée médiane des épisodes de diarrhée et de douleurs abdominales a été inférieure à 1 semaine. Aucune relation dose-effet n'a été observée s'agissant de la diarrhée ou des douleurs abdominales. Une suspension du traitement ou une réduction de la dose en raison d'effets indésirables gastro-intestinaux a eu lieu chez 4 patients atteints du syndrome d'Alagille (4,7 %) et chez 3 patients atteints de PFIC (6,4 %), et elles ont conduit à une amélioration ou une résolution des effets indésirables. Un patient atteint de PFIC (2,1 %) ayant présenté une diarrhée légère a arrêté le traitement ; en revanche, aucun patient n'a arrêté le traitement par Livmarli en raison d'effets indésirables gastro-intestinaux.
Si la diarrhée et/ou les douleurs abdominales persistent et qu'aucune autre étiologie n'est identifiée, une réduction de la dose ou une suspension du traitement devront être envisagées. La déshydratation doit être surveillée et rapidement traitée, le cas échéant. Si l'administration de Livmarli est suspendue, le traitement peut être redémarré à la dose tolérée une fois que la diarrhée ou les douleurs abdominales se sont améliorées (voir rubrique Posologie et mode d'administration).
Les augmentations des ALAT et ASAT, accompagnées pour certaines d'une augmentation de la bilirubine, ont été le plus souvent transitoires et d'intensité légère ou modérée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
ENVISAGER
un autre traitement si la réponse clinique adéquate n'est pas obtenue
chez le patient après 3 mois de traitement quotidien continu.
SURVEILLANCE du traitement :
- Bilan hépatique, AVANT et PENDANT le traitement.
- Taux de vitamines liposolubles (vitamines A, D, E) et le rapport
normalisé international (INR), AVANT d'instaurer le traitement et
PENDANT le traitement conformément aux pratiques cliniques usuelles.
Les patients atteints de PFIC2 chez lesquels la protéine BSEP (pompe
d'export des sels biliaires) est totalement absente ou dysfonctionnelle
(c.-à-d. les patients atteints du sous-type BSEP3 de la PFIC2) ne
devraient pas répondre au maralixibat.
PFIC
: en raison de la présence de propylène glycol, la dose maximale
recommandée, chez les patients atteints d'insuffisance rénale modérée
(clairance de la créatinine [ClCr] > 30 et < 60 mL/min), respectivement, hépatique modérée, est de 285 microgrammes/kg deux fois par jour.
Grossesse
Il n'existe pas de données sur l'utilisation du maralixibat chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité préclinique). Aucun effet sur le fœtus pendant la grossesse n'est attendu dans la mesure où l'exposition systémique au maralixibat est négligeable. Par mesure de précaution, il est préférable d'éviter l'utilisation de Livmarli pendant la grossesse.
Allaitement
Aucun effet sur les nouveau-nés/nourrissons allaités n'est attendu dans la mesure où l'exposition systémique de la femme qui allaite au maralixibat est négligeable. En raison de la présence de propylène glycol, par mesure de précaution, il est préférable d'éviter l'utilisation de Livmarli au cours de l'allaitement.
Fertilité
Aucune donnée clinique n'est disponible concernant les effets du maralixibat sur la fertilité. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets directs ou indirects sur la fertilité ou la reproduction (voir rubrique Données de sécurité préclinique).
Le maralixibat est un inhibiteur de l'OATP2B1, d'après les études in vitro. Une diminution de l'absorption orale des substrats de l'OATP2B1 (fluvastatine ou rosuvastatine, par exemple) due à l'inhibition de l'OATP2B1 dans le tube digestif ne peut être exclue. Une surveillance des effets des substrats de l'OATP2B1 devra être envisagée, si nécessaire.
Le maralixibat est également un inhibiteur du CYP3A4, d'après les études in vitro. Par conséquent, une augmentation des concentrations plasmatiques des substrats du CYP3A4 (midazolam, simvastatine, par exemple) ne peut être exclue et la prudence est recommandée en cas d'administration concomitante de telles substances.
Le maralixibat est un inhibiteur de l'absorption des acides biliaires, mais les interactions potentielles avec l'acide ursodésoxycholique (AUDC) biliaire n'ont pas été pleinement évaluées.
L'absorption du maralixibat est minime, sa métabolisation n'est pas significative et il n'est pas un substrat des transporteurs de substances actives ; par conséquent, aucun effet des autres médicaments concomitants sur l'élimination du maralixibat n'a été identifié.
Aucun effet inhibiteur ou inducteur du maralixibat sur les autres enzymes du cytochrome P450 n'a été mis en évidence chez les patients ; par conséquent, le maralixibat ne devrait pas affecter l'élimination des médicaments concomitants par le biais de ces mécanismes.
Le traitement par Livmarli doit être instauré sous la supervision d'un médecin expérimenté dans la prise en charge des patients atteints de maladies hépatiques cholestatiques.
Syndrome d'Alagille
La dose cible recommandée est de 380 microgrammes/kg une fois par jour. La dose initiale est de 190 microgrammes/kg une fois par jour et la dose devra être augmentée à 380 microgrammes/kg une fois par jour au bout d'une semaine. Le tableau 1 présente les doses à administrer (en mL de solution) en fonction du poids. Si le traitement est mal toléré, une réduction de la dose de 380 microgrammes/kg/jour à 190 microgrammes/kg/jour ou une interruption du traitement devront être envisagées. Une nouvelle augmentation de dose peut être envisagée en fonction de la tolérance. La dose quotidienne maximale recommandée en volume pour les patients pesant plus de 70 kg est de 3 mL (28,5 mg).
Tableau 1 : Volume de solution à administrer selon le poids du patient dans le syndrome d'Alagille
|
Poids du patient (kg) |
Jours 1 à 7 (190 microgrammes/kg une fois par jour) |
À partir du jour 8 (380 microgrammes/kg une fois par jour) |
||
|
Volume à administrer une fois par jour (mL) |
Taille de seringue pour administration orale (mL) |
Volume à administrer une fois par jour (mL) |
Taille de seringue pour administration orale (mL) |
|
|
5-6 |
0,1 |
0,5 |
0,2 |
0,5 |
|
7-9 |
0,15 |
0,3 |
||
|
10-12 |
0,2 |
0,45 |
||
|
13-15 |
0,3 |
0,6 |
1 |
|
|
16-19 |
0,35 |
0,7 |
||
|
20-24 |
0,45 |
0,9 |
||
|
25-29 |
0,5 |
1 |
||
|
30-34 |
0,6 |
1 |
1,25 |
3 |
|
35-39 |
0,7 |
1,5 |
||
|
40-49 |
0,9 |
1,75 |
||
|
50-59 |
1 |
2,25 |
||
|
60-69 |
1,25 |
3 |
2,5 |
|
|
70 ou plus |
1,5 |
3 |
||
Cholestase intrahépatique progressive familiale (PFIC)
La dose initiale est de 285 microgrammes/kg une fois par jour (1×/j) et elle peut être augmentée après 1 à 2 semaines à 285 microgrammes/kg deux fois par jour (2×/j, matin et soir). Après 1 à 2 semaines, la dose peut être encore augmentée à 570 microgrammes/kg deux fois par jour si cliniquement justifié et si le traitement est bien toléré. Le tableau 2 présente les doses à administrer (en mL de solution) en fonction du poids. Si le traitement est mal toléré, une réduction de la dose ou une interruption du traitement devront être envisagées. Une nouvelle augmentation de dose peut être envisagée si le traitement est suffisamment toléré. La dose quotidienne maximale recommandée pour les patients pesant plus de 50 kg est un volume de 6 mL (57 mg).
Tableau 2 : Volume de solution à administrer selon le poids du patient dans la PFIC
|
Poids du patient (kg) |
285 microgrammes/kg |
570 microgrammes/kg |
||
|
Volume à administrer 1×/j ou 2×/j (mL) |
Taille du dispensateur de dose (mL) |
Volume à administrer 2×/j (mL) |
Taille du dispensateur de dose (mL) |
|
|
3 |
0,1 |
0,5 |
0,2 |
0.5 |
|
4 |
0,1 |
0,25 |
||
|
5 |
0,15 |
0,3 |
||
|
6 à 7 |
0,2 |
0,4 |
||
|
8 à 9 |
0,25 |
0,5 |
||
|
10 à 12 |
0,35 |
0,6 |
1 |
|
|
13 à 15 |
0,4 |
0,8 |
||
|
16 à 19 |
0,5 |
1 |
||
|
20 à 24 |
0,6 |
1 |
1,25 |
3 |
|
25 à 29 |
0,8 |
1,5 |
||
|
30 à 34 |
0,9 |
2 |
||
|
Poids du patient (kg) |
285 microgrammes/kg |
570 microgrammes/kg |
||
|
Volume à administrer 1×/j ou 2×/j (mL) |
Taille du dispensateur de dose (mL) |
Volume à administrer 2×/j (mL) |
Taille du dispensateur de dose (mL) |
|
|
35 à 39 |
1,25 |
3 |
2,25 |
|
|
40 à 49 |
1,25 |
2,75 |
||
|
50 à 59 |
1,5 |
3 |
||
|
60 à 69 |
2 |
3 |
||
|
70 à 79 |
2,25 |
3 |
||
|
80 ou plus |
2,5 |
3 |
||
Le recours à un autre traitement devra être envisagé si la réponse clinique adéquate n'est pas obtenue chez le patient après 3 mois de traitement quotidien continu par le maralixibat.
Oubli d'une dose
En cas d'oubli, il conviendra de renoncer à la dose oubliée et de poursuivre le traitement normalement au moment prévu pour la prise suivante.
Populations particulières
Insuffisance rénale
Le maralixibat n'a pas été étudié chez les patients atteints d'insuffisance rénale ou d'insuffisance rénale terminale (IRT) nécessitant une hémodialyse. Les concentrations plasmatiques du maralixibat sont minimes et son excrétion rénale est négligeable (voir rubrique Propriétés pharmacocinétiques).
Syndrome d'Alagille : aucun ajustement posologique n'est nécessaire.
PFIC : en raison de la présence de propylène glycol, la dose maximale recommandée de Livmarli chez les patients atteints d'insuffisance rénale modérée (clairance de la créatinine [ClCr] ≥ 30 et < 60 mL/min) est de 285 microgrammes/kg deux fois par jour. Livmarli ne doit pas être utilisé chez les patients atteints de PFIC et d'insuffisance rénale sévère (ClCr < 30 mL/min ; voir rubriques Contre-indications et Mises en garde spéciales et précautions d'emploi).
Insuffisance hépatique
Le maralixibat n'a pas été suffisamment étudié chez les patients atteints d'insuffisance hépatique. Syndrome d'Alagille : compte tenu de l'absorption minime du maralixibat, aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique. Une étroite surveillance est toutefois conseillée chez les patients présentant une insuffisance hépatique terminale ou en cas de décompensation hépatique.
PFIC : en raison de la présence de propylène glycol, la dose maximale recommandée de Livmarli chez les patients atteints d'insuffisance hépatique modérée est de 285 microgrammes/kg deux fois par jour. Livmarli ne doit pas être utilisé chez les patients atteints de PFIC et d'insuffisance hépatique sévère (voir rubriques Contre-indications et Mises en garde spéciales et précautions d'emploi).
Population pédiatrique
La sécurité et l'efficacité de Livmarli chez les enfants atteints du syndrome d'Alagille âgés de moins de 2 mois et chez les enfants atteints de PFIC âgés de moins de 3 mois n'ont pas été établies. Les données actuellement disponibles sont décrites aux rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques mais aucune recommandation sur la posologie ne peut être donnée pour ces groupes d'âge. Syndrome d'Alagille (≥ 2 mois) : aucun ajustement posologique n'est nécessaire.
PFIC (≥ 3 mois) : en raison de la présence de propylène glycol, la dose maximale recommandée de Livmarli chez les patients atteints de PFIC âgés de moins de 5 ans est de 285 microgrammes/kg deux fois par jour (voir rubrique Mises en garde spéciales et précautions d'emploi).
Il convient de prêter une attention particulière au calcul précis de la dose de Livmarli et de communiquer clairement les instructions posologiques aux aidants et aux patients afin de limiter le risque d'erreur de dosage et de surdosage.
Mode d'administration
Livmarli doit être administré par voie orale par un aidant ou par le patient à l'aide d'une seringue pour administration orale, avant un repas (au plus dans les 30 minutes précédentes) ou au cours d'un repas, le matin en cas d'administration une fois par jour, ou le matin et le soir en cas d'administration deux fois par jour.
L'utilisation de la solution buvable de Livmarli mélangée directement à un aliment ou une boisson avant administration n'a pas été étudiée et doit être évitée.
Des seringues pour administration orale de trois tailles (0,5 mL, 1 mL et 3 mL) sont fournies avec chaque flacon de Livmarli. Les tableaux 1 et 2 indiquent la taille de seringue pour administration orale adaptée en fonction du poids du patient.
Durée de conservation :
30 mois.
Après première ouverture
Après la première ouverture du flacon, le médicament doit être conservé à une température ne dépassant pas 30 °C et être utilisé dans les 130 jours. Passé ce délai, le flacon et son contenu doivent être éliminés, même si le flacon n'est pas vide.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température. À conserver dans l'emballage d'origine, à l'abri de la lumière.
Pour les conditions de conservation du médicament après première ouverture, voir la rubrique Durée de conservation.
Sans objet.
L'absorption du maralixibat au niveau du tube digestif est minime et le surdosage ne devrait pas aboutir à des concentrations plasmatiques élevées de la substance active. Des doses uniques allant jusqu'à 500 mg, soit environ 18 fois la dose recommandée, ont été administrées à des volontaires sains adultes, sans conséquences délétères.
Livmarli contient du propylène glycol ; le surdosage du médicament pourrait conduire à un surdosage en propylène glycol (voir rubrique Mises en garde spéciales et précautions d'emploi).
En cas de surdosage, il conviendra de surveiller les signes ou symptômes de toxicité du propylène glycol chez le patient (voir rubrique Mises en garde spéciales et précautions d'emploi) et de mettre en place une prise en charge adaptée. En cas de surdosage, le propylène glycol peut être éliminé de l'organisme par dialyse.
Classe pharmacothérapeutique : traitement de la bile et du foie, autres médicaments pour le traitement de la bile, Code ATC : A05AX04
Mécanisme d'action
Le maralixibat est un inhibiteur sélectif, puissant et réversible du transporteur iléal des acides biliaires (IBAT), faisant l'objet d'une absorption minime.
Le maralixibat agit localement, dans la section distale de l'iléon, en réduisant la recapture des acides biliaires et en augmentant la clairance des acides biliaires par le biais du côlon, diminuant ainsi la concentration des acides biliaires dans le sérum.
Efficacité clinique dans le syndrome d'Alagille
L'efficacité du maralixibat chez les patients atteints du syndrome d'Alagille a été évaluée dans une étude de 48 semaines comprenant une période d'introduction de la substance active en ouvert sur 18 semaines, une période de retrait de traitement randomisé en double aveugle sur 4 semaines et une période d'extension en ouvert à long terme.
Trente-et-un (31) patients atteints du syndrome d'Alagille et présentant une cholestase et un prurit ont été inclus, parmi lesquels 90,3 % recevaient au moins un médicament pour le traitement du prurit lors de leur entrée dans l'étude (traitement par la rifampicine et par l'acide ursodésoxycholique chez 74,2 % et 80,6 % des patients, respectivement). L'utilisation concomitante de ces médicaments était autorisée pendant l'étude, mais les ajustements posologiques étaient interdits au cours des 22 premières semaines. Tous les patients présentaient un syndrome d'Alagille dû à la mutation JAGGED1.
Les critères d'exclusion comprenaient l'interruption chirurgicale du cycle entéro-hépatique, les antécédents ou la présence actuelle de toute affection connue pour interférer avec l'absorption, la distribution, le métabolisme ou l'excrétion des médicaments, y compris le métabolisme des sels biliaires dans l'intestin, et la diarrhée chronique nécessitant une réhydratation intraveineuse ou une intervention nutritionnelle.
Après une période initiale d'escalade de dose de 5 semaines, les patients ont reçu un traitement en ouvert par le maralixibat à la dose de 380 microgrammes/kg une fois par jour pendant 13 semaines ; deux patients ont arrêté le traitement au cours de ces 18 premières semaines d'introduction du traitement en ouvert. Les 29 patients ayant terminé la phase d'introduction en ouvert ont ensuite été randomisés en vue de poursuivre le traitement par le maralixibat ou de recevoir le placebo correspondant (n = 16 pour le placebo, n = 13 pour le maralixibat) pendant les 4 semaines de la période de retrait de traitement randomisé en double aveugle (semaines 19-22). Ces 29 patients sont tous allés au terme de la période de retrait de traitement randomisé en aveugle ; tous les patients ont ensuite reçu le maralixibat en ouvert à la dose de 380 microgrammes/kg une fois par jour jusqu'à la semaine 48. Pour les patients qui recevaient précédemment le placebo, une escalade de dose a alors été réalisée suivant le même schéma que lors de la période initiale.
L'âge médian des patients randomisés était de 5 ans (intervalle : 1 à 15 ans) et 66 % étaient de sexe masculin. À l'inclusion, les valeurs moyennes (écart type [ET]) des paramètres du bilan hépatique étaient les suivantes : taux d'acides biliaires sériques (ABs) 280 (213) µmol/L, aspartate aminotransférase (ASAT) 158 (68) U/L, alanine aminotransférase (ALAT) 179 (112) U/L, gamma glutamyl-transférase (GGT) 498 (399) U/L et bilirubine totale (BT) 5,6 (5,4) mg/dL.
Taux d'acides biliaires sériques (ABs)
Par rapport à l'inclusion, une réduction moyenne (ET) statistiquement significative du taux d'ABs a été observée, à hauteur de 88 (120) et 96 (166,6) μmol/L respectivement aux semaines 18 et 48, lorsque les patients recevaient le maralixibat. À l'issue de la période contrôlée contre placebo, une différence statistiquement significative de moyenne des moindres carrés (ES) a été mise en évidence entre les groupes maralixibat et placebo s'agissant de l'évolution du taux d'ABs entre la semaine 18 et la semaine 22 (‑114 [48,0] µmol/L ; p = 0,025). Lorsque le groupe placebo a repris le traitement par le maralixibat, à l'issue de la période de retrait de traitement, les taux d'ABs sont redescendus au niveau précédemment observé sous traitement par le maralixibat (voir la figure 1).
Figure 1 : Évolution moyenne (± ES) du taux d'ABs sur 48 semaines par rapport à l'inclusion, chez l'ensemble des patients
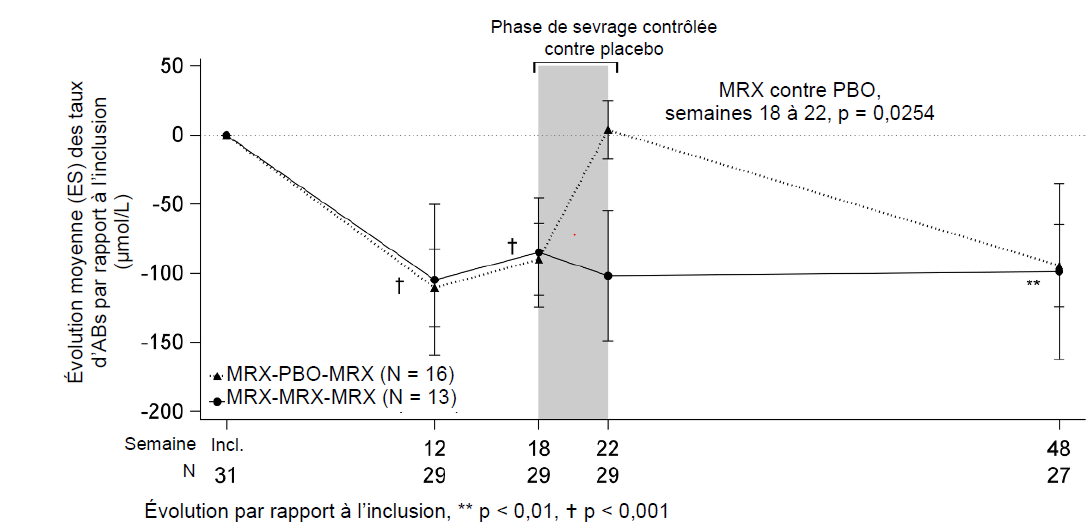
MRX = maralixibat ; PBO = placebo ; ES = erreur standard ; Incl. = inclusion
Prurit
La sévérité du prurit a été évaluée au sein de la population globale (n = 31) et mesurée d'après le score ItchRO(Obs) (Itch Reported Outcome Observer). Le score ItchRO est établi par les aidants sur une échelle validée allant de 0 à 4 (0 = aucun ; 4 = très sévère) et il a été montré que des modifications ≥ 1,0 sont cliniquement significatives. L'évolution de la sévérité du prurit chez les participants traités par le maralixibat par comparaison avec ceux traités par le placebo pendant la période de retrait de traitement randomisé, ainsi que son évolution entre l'inclusion et les semaines 18 et 48 ont été mesurées. Le score ItchRO(Obs) moyen à l'inclusion était de 2,9.
Chez les patients ayant reçu le maralixibat, une évolution cliniquement significative et des réductions statistiquement significatives du score ItchRO(Obs) ont été observées par rapport à l'inclusion, à savoir ‑1,7 et ‑1,6 points, respectivement, à la semaine 18 et à la semaine 48.
Pendant la période de retrait de traitement randomisé contrôlé contre placebo, la réduction du prurit s'est maintenue chez les patients traités par le maralixibat, tandis que les scores de prurit sont revenus à leur niveau initial chez les patients du groupe placebo. La différence de moyenne des moindres carrés (ES) entre les groupes maralixibat et placebo s'agissant de l'évolution du prurit entre les semaines 18 et 22 (‑1,5 [0,3] ; IC à 95 % : ‑2,1 à ‑0,8 ; p < 0,0001 ; voir la figure 2) a été statistiquement significative. Après reprise du traitement par maralixibat, le score de prurit des patients du groupe placebo s'est à nouveau amélioré lors de la semaine 28. Chez les patients traités par le maralixibat, la réduction du prurit s'est maintenue jusqu'à la semaine 48.
Figure 2 : Évolution de la moyenne hebdomadaire du score de sévérité ItchRO(Obs) mesuré le matin par rapport à l'inclusion, sur 48 semaines, en fonction du groupe de randomisation, chez l'ensemble des patients
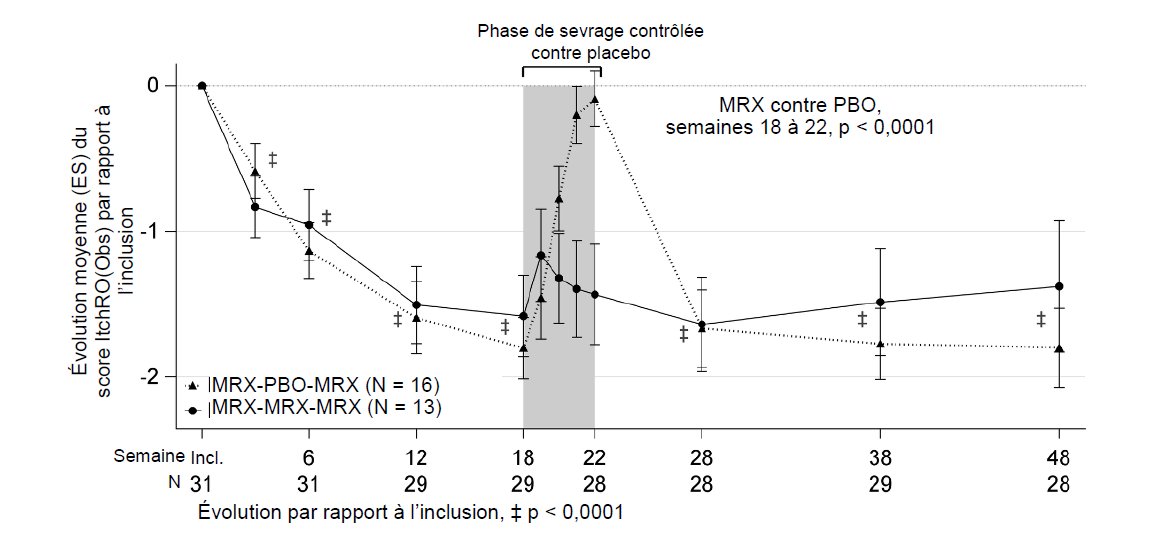
MRX = maralixibat ; PBO = placebo ; ES = erreur standard ; Incl. = inclusion
Pour le cholestérol et la sévérité des xanthomes, des améliorations d'ampleur variable ont été observées au cours du traitement par le maralixibat.
Le mécanisme d'action du maralixibat conduisant à la recapture des acides biliaires devrait être similaire dans toutes les tranches d'âge. Les données démontrant l'efficacité chez les patients âgés de moins de 12 mois atteints du syndrome d'Alagille sont limitées. Au cours d'une étude en ouvert, avec groupe unique, menée chez 8 patients âgés de 2 à 10 mois atteints du syndrome d'Alagille, l'évolution moyenne (ET ; médiane ; intervalle) du score de prurit sur l'échelle de grattage évalué par le médecin (Clinician Scratch Scale) (allant de 0 = aucun à 4 = mutilation cutanée, hémorragie et cicatrices manifestes) à la semaine 13 a été de ‑0,2 (1,91 ; ‑1,0 ; ‑3,0 à 3,0) et l'évolution moyenne (ET ; médiane ; intervalle) du taux d'ABs a été de ‑88,91 µmol/L (113,348 ; ‑53,65 ; ‑306,1 à 14,4). Chez deux patients, le prurit et le taux d'ABs se sont tous deux améliorés.
Efficacité clinique dans la PFIC
L'efficacité du maralixibat a été évaluée dans une étude randomisée, en double aveugle, contrôlée contre placebo, sur une durée de 26 semaines (MRX‑502). Au total, 93 patients diagnostiqués PFIC sur la base d'une cholestase intrahépatique avec prurit persistant, anomalies des paramètres du bilan hépatique et/ou mise en évidence d'une atteinte hépatique progressive et âgés de plus de 12 mois et de moins de 18 ans, ont été inclus dans l'étude. Un génotypage a été réalisé pour confirmer le type de PFIC. Le prurit persistant était défini comme un prurit d'une durée > 6 mois avec un score de prurit moyen sur l'échelle ItchRO[Obs] supérieur ou égal à 1,5 dans les 4 semaines précédant l'inclusion.
Les patients étaient exclus de l'étude en cas de cirrhose décompensée, d'antécédents ou de présence actuelle de toute affection connue pour interférer avec l'absorption, la distribution, le métabolisme ou l'excrétion des médicaments, y compris le métabolisme des sels biliaires dans l'intestin, ou de diarrhée chronique nécessitant l'administration de liquides en intraveineuse ou une intervention nutritionnelle.
Les patients ont été randomisés selon un ratio de 1:1 en vue de recevoir 570 microgrammes/kg de maralixibat (n = 47) ou un placebo par voie orale (n = 46) deux fois par jour pendant 26 semaines, avec une période d'escalade de dose de 4 à 6 semaines en débutant à la dose de 142 microgrammes/kg deux fois par jour. Au total, 92,5 % des patients (44/47 sous maralixibat et 42/46 sous placebo) ont terminé la période d'étude de 26 semaines, 7 patients ayant arrêté l'étude (4 retraits du consentement, 1 EI de diarrhée légère, 1 transplantation hépatique et 1 progression de la maladie). Les patients ayant terminé l'étude pivot étaient éligibles pour participer à l'étude d'extension en ouvert (MRX‑503).
Les critères d'efficacité de l'étude pivot comprenaient l'évolution de la sévérité du prurit, des taux d'acides biliaires sériques, des paramètres du bilan hépatique et de la croissance.
Les critères d'efficacité ont été évalués chez les patients dont les résultats de test génétique concordaient avec les variants bi-alléliques responsables de la PFIC (n = 64) : ABCB11/BSEP (PFIC2) n = 31 ; ATP8B1/FIC1 (PFIC1) n = 13 ; ABCB4/MDR3 (PFIC3) n = 9 ; TJP2 (PFIC4) n = 7 ; MYO5B(PFIC6) n = 4. Il y avait davantage de sujets de sexe féminin (53,1 %) et l'âge moyen était de 4,6 ans (intervalle : 1 à 15 ans). La plupart des patients recevaient un traitement stable par l'acide ursodésoxycholique (89,1 %) ou la rifampicine (51,6 %) à l'inclusion. Les valeurs moyennes (écart type [ET]) des paramètres du bilan hépatique à l'inclusion étaient les suivantes : taux d'acides biliaires sériques de 263 (143) μmol/L, ASAT de 113 (82) U/L, ALAT de 107 (87) U/L, bilirubine totale (BT) de 69,8 (70,1) µmol/L, bilirubine directe (BD) de 50,6 (52,4) µmol/L. La moyenne (ET) des scores de sévérité matinale du prurit sur l'échelle ItchRO[Obs] à l'inclusion était de 2,8 (0,87). Il n'y avait aucune différence significative entre les groupes de traitement s'agissant des caractéristiques initiales ou des paramètres liés à la maladie.
Taux d'acides biliaires sériques (ABs)
La différence entre les groupes de traitement par maralixibat et par placebo sur l'évolution moyenne du taux d'acides biliaires sériques total entre l'inclusion et la moyenne des semaines 18, 22 et 26 a été statistiquement significative, avec une variation de moyenne des moindres carrés (MMC) par rapport au placebo de -160 µmol/L (IC à 95 % : -220,8 ; -100,0) (figure 3).
Figure 3 : Évolution observée des taux d'acides biliaires sériques moyens au fil du temps dans la PFIC de type 1, 2, 3, 4 et 6 (étude MRX-502)
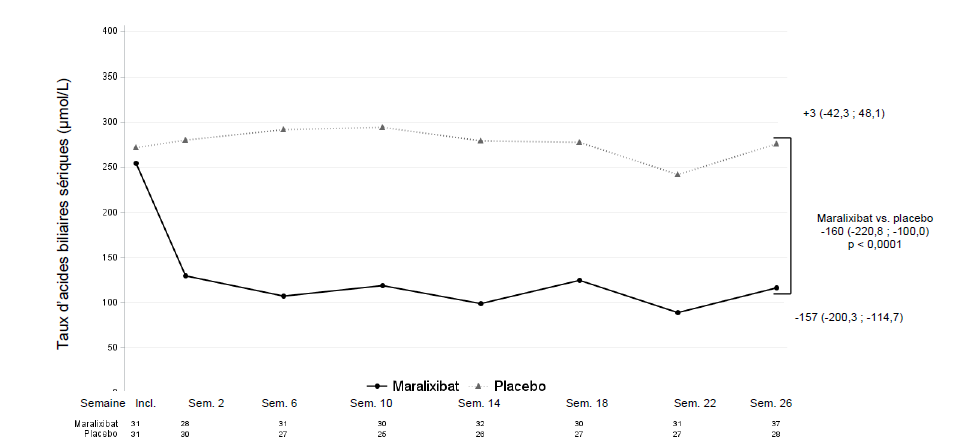
Incl. = inclusion ; Sem. = semaine. Les valeurs observées sont affichées. Les statistiques présentées sont les moyennes des semaines 18, 22 et 26 calculées en utilisant une pondération égale des 3 estimations spécifiques à chaque visite obtenues à l'aide d'un modèle mixte pour mesures répétées (MMMR), avec l'évolution par rapport à l'inclusion comme variable dépendante et en utilisant comme effets catégoriels fixes le groupe de traitement, le type de PFIC, la visite d'analyse et l'interaction traitement/visite, ainsi que les covariables fixes continues du score initial et de l'interaction score initial/visite. L'estimation de la moyenne des moindres carrés et l'intervalle de confiance à 95 % sont présentés.
Le pourcentage de répondeurs pour le taux d'acides biliaires sériques a été de 45,5 % avec le maralixibat contre 6,5 % sous placebo, soit une différence (IC à 95 %) de 39,0 % (16,5 % ; 58,2 %). Les participants étaient considérés comme répondeurs pour le taux d'acides biliaires sériques s'ils présentaient un taux d'ABs moyen < 102 µmol/L (valable uniquement si le taux d'ABs à l'inclusion était ≥ 102 µmol/L) OU une réduction moyenne ≥ 75 % par rapport à l'inclusion. Pour déterminer la réponse, les valeurs moyennes des taux d'ABs aux semaines 18, 22 et 26 ont été utilisées.
Prurit
Une différence entre les groupes de traitement par maralixibat et par placebo a été démontrée en faveur du maralixibat s'agissant de l'évolution moyenne du score de sévérité matinale du prurit sur l'échelle ItchRO(Obs) entre l'inclusion et les semaines 15-26, avec une variation de MMC par rapport au placebo de -1,200 (IC à 95 % : -1,727 ; -0,674 ; figure 4).
Figure 4 : Moyenne hebdomadaire observée des scores de prurit matinaux quotidiens au fil du temps dans la PFIC de type 1, 2, 3, 4 et 6 (étude MRX-502)
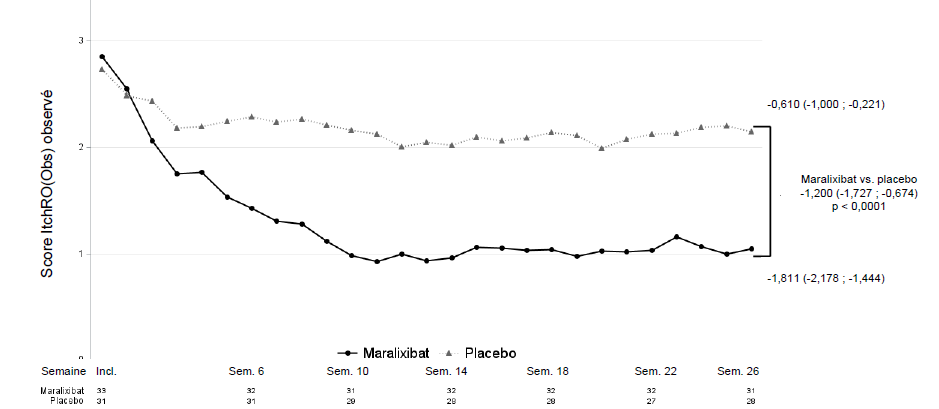
Incl. = inclusion ; Sem. = semaine. Les valeurs observées sont affichées. Les statistiques présentées sont les moyennes des périodes des semaines 15-18, 19-22 et 23-26 calculées en utilisant une pondération égale des 3 estimations spécifiques à chaque visite obtenues à l'aide d'un modèle mixte pour mesures répétées (MMMR), avec l'évolution par rapport à l'inclusion comme variable dépendante et en utilisant comme effets catégoriels fixes le groupe de traitement, le type de PFIC, la visite d'analyse et l'interaction traitement/visite, ainsi que les covariables fixes continues du score initial et de l'interaction score initial/visite. L'estimation de la moyenne des moindres carrés et l'intervalle de confiance à 95 % sont présentés.
Le tableau 4 présente les résultats de la comparaison des scores ItchRO(Obs) entre le maralixibat et le placebo.
Tableau 4 : Proportion de répondeurs pour le prurit (étude MRX-502)

Les valeurs de p pour la comparaison entre les groupes de traitement par maralixibat et par placebo ont été calculées en utilisant un test exact de Barnard.
Les intervalles de confiance exacts à 95 % sont basés sur une statistique score.
Les analyses exploratoires ont montré une réduction plus importante (amélioration) des scores moyens de troubles du sommeil dans le groupe traité par le maralixibat par comparaison avec le groupe placebo. Les analyses exploratoires ont montré des améliorations de la bilirubine durant le traitement par le maralixibat (tableau 5). Les taux de bilirubine totale qui étaient anormaux à l'inclusion s'étaient normalisés à la semaine 26 chez 40 % (10/25) des patients sous maralixibat, contre 0 % (0/18) des patients sous placebo. Une augmentation plus importante (amélioration) du z-score de poids a été observée dans le groupe traité par le maralixibat par comparaison avec le placebo (variation de MMC par rapport au placebo : 0,227 [IC à 95 % : 0,012 ; 0,442 ; tableau 5]).
Tableau 5 : Paramètres du bilan hépatique et paramètres de croissance pour le maralixibat vs. placebo sur la période de traitement de 26 semaines chez les participants atteints de PFIC dans l'étude pivot (MRX-502, analyses exploratoires)
|
Critère d'efficacité |
Placebo (n = 31) |
Maralixibat (n = 33) |
|
Alanine aminotransférase (U/L) |
|
|
|
Inclusion (moyenne [ES]) |
127,3 (18,68) |
87,8 (10,77) |
|
Variation de MMC entre l'incl. [ES] et les semaines 18-26 |
-7,0 (11,13) |
9,7 (10,36) |
|
Différence de MMC par rapport au placebo (IC à 95 %)
|
|
16,6 (-13,31 ; 46,60) |
|
Aspartate aminotransférase (U/L) |
|
|
|
Inclusion (moyenne [ES]) |
129,8 (18,12) |
96,9 (9,57) |
|
Variation de MMC entre l'incl. [ES] et les semaines 18-26 |
-0,4 (14,91) |
13,6 (14,05) |
|
Différence de MMC par rapport au placebo (IC à 95 %)
|
|
14,1 (-26,57 ; 54,69) |
|
Bilirubine totale (µmol/L) |
|
|
|
Inclusion (moyenne [ES]) |
69,1 (13,69) |
70,4 (11,32) |
|
Variation de MMC entre l'incl. [ES] et les semaines 18-26 |
15,9 (12,37) |
-18,3 (11,65) |
|
Différence de MMC par rapport au placebo (IC à 95 %)
|
|
-34,3 (-68,06 ; -0,46) |
|
Bilirubine directe (µmol/L) |
|
|
|
Inclusion (moyenne [ES]) |
50,2 (10,28) |
50,9 (8,40) |
|
Variation de MMC entre l'incl. [ES] et les semaines 18-26 |
13,5 (9,52) |
-12,9 (8,97) |
|
Différence de MMC par rapport au placebo (IC à 95 %)
|
|
-26,4 (-52,46 ; -0,26) |
|
z-score de taille |
|
|
|
Inclusion (moyenne [ES]) |
-2,06 (0,27) |
-2,08 (0,23) |
|
Variation de MMC entre l'incl. [ES] et les semaines 18-26 |
-0,13 (0,09) |
0,08 (0,09) |
|
Différence de MMC par rapport au placebo (IC à 95 %)
|
|
0,21 (-0,04 ; 0,5) |
|
z-score de poids |
|
|
|
Inclusion (moyenne [ES]) |
-1,28 (0,24) |
-1,75 (0,23) |
|
Variation de MMC entre l'incl. [ES] et les semaines 18-26 |
0,12 (0,08) |
0,35 (0,07) |
|
Différence de MMC par rapport au placebo (IC à 95 %)
|
|
0,23 (0,01 ; 0,4) |
ES = erreur standard ; MMC = moyenne des moindres carrés ; IC = intervalle de confiance ; incl. = inclusion. Les valeurs à l'inclusion sont des valeurs observées. Les valeurs de MMC sont des moyennes des semaines 18, 22 et 26 calculées en utilisant une pondération égale des 3 estimations spécifiques à chaque visite obtenues à l'aide d'un modèle mixte pour mesures répétées (MMMR), avec l'évolution par rapport à l'inclusion comme variable dépendante et en utilisant comme effets catégoriels fixes le groupe de traitement, le type de PFIC, la visite d'analyse et l'interaction traitement/visite, ainsi que les covariables fixes continues du score initial et de l'interaction score initial/visite.
Parmi les 64 patients de l'étude pivot (MRX‑502) dont les résultats de test génétique concordaient avec les variants bi-alléliques responsables de la PFIC, 57 ont été inclus dans une analyse intermédiaire de l'étude d'extension en ouvert qui est en cours (MRX‑503). La durée médiane du traitement par le maralixibat chez ces patients était de 47,3 semaines (intervalle : 4,1 semaines - 119,4 semaines). Un maintien des effets du traitement par le maralixibat sur les taux d'acides biliaires sériques et les taux de bilirubine ainsi que sur le prurit a été mis en évidence. Une amélioration supplémentaire des z-scores de taille et de poids a été observée.
Lors d'une étude de sécurité en ouvert, avec groupe unique (MRX‑801) menée chez 10 patients âgés de 1 à 11 mois et atteints de PFIC (le prurit actif n'était pas un critère requis), une diminution des taux d'ABs, de bilirubine totale et de bilirubine directe a été observée chez certains patients à la semaine 13. Deux patients ont également présenté une amélioration de leur prurit.
Circonstances exceptionnelles
Une autorisation de mise sur le marché « sous circonstances exceptionnelles » a été délivrée pour ce médicament. Cela signifie qu'en raison de la rareté de cette maladie, il n'a pas été possible d'obtenir des informations complètes concernant ce médicament. L'Agence européenne du médicament réévaluera chaque année toute nouvelle information qui pourrait être disponible, et, si nécessaire, ce RCP sera mis à jour.
Absorption
La cible du maralixibat se situe dans la lumière de l'intestin grêle, si bien que le passage du maralixibat dans le plasma n'est ni pertinent ni nécessaire à son efficacité. Le maralixibat fait l'objet d'une absorption minime et les concentrations plasmatiques sont souvent inférieures au seuil de détection (0,25 ng/mL) après administration de doses uniques ou multiples à la posologie thérapeutique. Sa biodisponibilité absolue est estimée à moins de 1 %.
Effets des aliments
L'absorption du maralixibat est relativement plus élevée lorsqu'il est administré à jeun, et aucun ajustement posologique n'est nécessaire sur la base de l'effet des aliments. Le maralixibat peut être pris avant un repas (au plus dans les 30 minutes précédentes) ou au cours d'un repas (voir rubrique Posologie et mode d'administration).
Distribution
Le maralixibat montre un taux de liaison élevé (91 %) avec les protéines plasmatiques humaines in vitro.
Lors d'un test ADME clinique avec administration de [14C] maralixibat, la radioactivité circulante est restée inférieure au seuil de détection lors de toutes les mesures. Aucune accumulation apparente du maralixibat n'a été notée.
Biotransformation
Aucun métabolite n'a été détecté dans le plasma et le maralixibat est métabolisé de façon minime dans le tube digestif.
Élimination
Le maralixibat est principalement éliminé dans les selles sous forme de substance mère non métabolisée, 0,066 % de la dose administrée étant excrétés dans les urines.
Populations particulières
Aucune différence cliniquement significative n'a été observée au niveau de la pharmacocinétique du maralixibat en fonction de l'âge, du sexe ou de l'origine ethnique.
Insuffisance hépatique
Des patients atteints du syndrome d'Alagille ou de PFIC et présentant un certain degré d'insuffisance hépatique ont été inclus dans les études cliniques sur le maralixibat. La majorité des patients présentaient un certain degré d'insuffisance hépatique selon la classification NCI‑ODWG en raison de la maladie. Toutefois, il n'est pas clairement établi à ce jour si cette classification est adaptée, dans le cadre de la maladie cholestatique, pour prédire l'influence sur la pharmacocinétique du maralixibat. Le maralixibat fait l'objet d'une absorption minime et les données obtenues chez l'animal indiquent que les très faibles concentrations plasmatiques sont dues à cette faible absorption et non à un effet de premier passage hépatique. Par ailleurs, les concentrations plasmatiques du maralixibat n'ont pas augmenté chez les patients qui présentaient une insuffisance hépatique selon les critères NCI‑ODWG. Cependant, la pharmacocinétique du maralixibat n'a pas fait l'objet d'une évaluation systématique chez les patients catégorisés selon la classification de Child‑Pugh (patients présentant une cirrhose et des signes de décompensation).
Insuffisance rénale
La pharmacocinétique du maralixibat n'a pas été étudiée chez les patients présentant une insuffisance rénale, y compris ceux atteints d'IRT ou ceux sous hémodialyse. Cependant, l'insuffisance rénale ne devrait pas influer sur la pharmacocinétique du maralixibat compte tenu de la faible exposition systémique et de l'excrétion urinaire quasiment nulle.
Livmarli n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études de pharmacologie de sécurité, pharmacologie secondaire, toxicologie en administration répétée, génotoxicité, cancérogénicité, fertilité, toxicité sur les fonctions de reproduction et de développement, et toxicité chez l'animal juvénile, n'ont pas révélé de risque particulier pour l'homme.
Les seringues pour administration orale doivent être rincées à l'eau, séchées à l'air libre et peuvent être réutilisées pendant 130 jours.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription initiale hospitalière
Prescription initiale réservée à certains spécialistes
Renouvellement de la prescription réservé aux spécialistes en HEPATO/GASTRO-ENTEROLOGIE
Renouvellement de la prescription réservé aux spécialistes en PEDIATRIE
Solution buvable.
Liquide limpide, incolore à jaune pâle.
Flacon de 30 mL en PET de couleur ambrée, muni d'un adaptateur en PEBD préinstallé et d'une fermeture de sécurité enfant en PEHD avec une doublure en mousse, contenant 30 mL de solution buvable.
Présentation :
Chaque boîte contient un flacon de 30 mL accompagné de trois seringues pour administration orale réutilisables (0,5 mL, 1 mL et 3 mL) portant les graduations suivantes :
Seringue de 0,5 mL en polypropylène avec un piston blanc : nombres inscrits tous les 0,1 mL, principaux traits de graduation tous les 0,05 mL et traits de graduation secondaires tous les 0,01 mL.
Seringue de 1 mL en polypropylène avec un piston blanc : nombres inscrits tous les 0,1 mL.
Seringue de 3 mL en polypropylène avec un piston blanc : nombres inscrits tous les 0,5 mL et traits de graduation tous les 0,25 mL entre 0,5 mL et 3 mL.
Chaque millilitre de solution contient l'équivalent de 9,5 mg de maralixibat sous la forme de chlorure de maralixibat.
Excipient à effet notoire :
Chaque millilitre de solution buvable contient 364,5 mg de propylène glycol (E 1520).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Propylène glycol (E 1520)
Édétate disodique
Sucralose
Arôme raisin
Eau purifiée